
What is cystic fibrosis?
Image credit: Keith Chambers / via Wikimedia Commons

Cystic fibrosis is a relatively common genetic condition in which the lungs and digestive system become clogged with thick, sticky mucus.
- Cystic fibrosis is a recessive genetic condition caused by mutations in the CFTR gene on chromosome 7.
- This can lead to chronic breathing difficulties and recurring lung infections, caused by the build-up of sticky mucus in the lungs and respiratory tubes. Long-term, this can cause lung scarring, called fibrosis.
What is cystic fibrosis?
- Cystic fibrosis is caused by various mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene on chromosome 7.
- The most common mutation involves the deletion of just three DNA bases (a single codon) from the CFTR gene.
- Cystic fibrosis is a recessive genetic disease, which means that both copies of a person’s CFTR gene must contain the mutation for cystic fibrosis to occur.
- If an individual has just one copy of the mutated gene they are said to be a carrier of the condition. About 1 in every 25 people is a carrier for cystic fibrosis.
- Cystic fibrosis has the highest incidence of any recessive disease in Caucasian populations (approximately 1 in 2,000) but is less common in other ethnic groups.
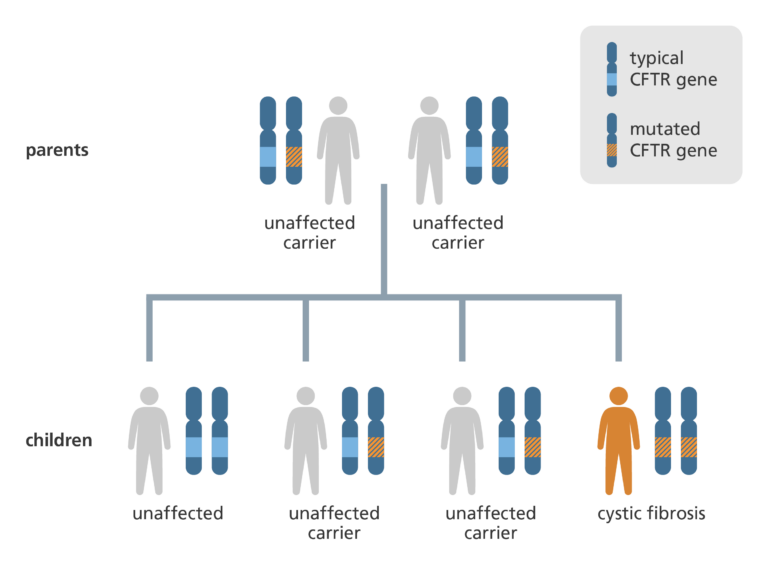
What is the biology of cystic fibrosis?
- The CFTR gene provides instructions for making a protein called the cystic fibrosis transmembrane conductance regulator (CFTR).
- The CFTR protein acts as a channel across the membrane of cells that are specialised to produce mucus, sweat, saliva, tears and digestive enzymes.
- This protein channel normally transports chloride ions in and out of these cells which helps control the movement of water in tissues, ensuring that the mucus in our airways and various body systems remains thin and free-flowing.
- However, changes to the CFTR protein can disrupt its function as a chloride channel, inhibiting the flow of chloride ions and water in and out of the cells.
- This means mucus-producing cells secrete mucus that is abnormally thick and sticky, which can obstruct the airways and glands, resulting in the symptoms of cystic fibrosis.
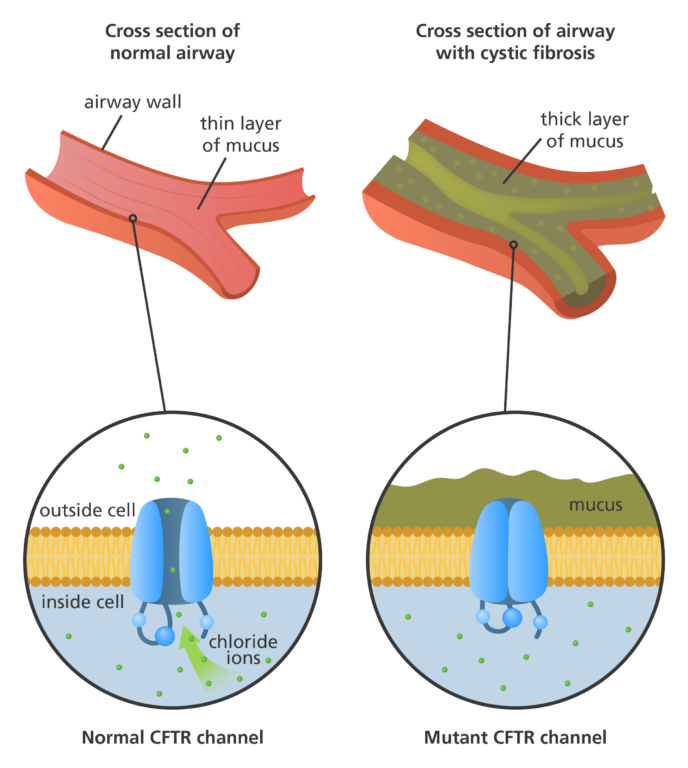
What are the symptoms of cystic fibrosis?
- The most common symptoms of cystic fibrosis are coughing, chronic breathing difficulties and recurrent lung infections, caused by the build-up of sticky mucus in the lungs and respiratory tubes.
- In the long term this can result in lung scarring, called fibrosis, damaging the airways of the lungs. In turn, this leads to a tendency toward more infections and decreased lung function.
- Around 85% of people with cystic fibrosis also have a deficiency in their pancreas caused by thick mucus blocking the pancreatic ducts. This prevents the pancreas from delivering enzymes needed for digestion and from producing insulin, which can lead to diabetes. Children with this aspect of the disease can suffer from malnutrition and struggle to put on weight.
- Cystic fibrosis can also cause infertility, because the ‘vasa deferetia’ – the tubes that transport sperm – are usually missing.
How is cystic fibrosis diagnosed?
- In families where there is a history of cystic fibrosis, a prenatal test can be offered, looking for the mutated form of the CFTR gene.
- In the UK, all newborn babies are screened for cystic fibrosis. The genetic test can also be used to diagnose cystic fibrosis in older children and adults who didn’t have the newborn test.
- Without a family history, clues to the disease can include sweat tasting very salty, persistent lung infections and a slow growth rate. A simple test that measures the amount of salt in sweat can be used to diagnose the disease.
How is cystic fibrosis managed?
- There is no cure for cystic fibrosis but there are therapies and medicines that can help control the symptoms and prevent or reduce the risk of complications.
- Daily physiotherapy can help patients with the condition. Drumming and patting on the patient’s back and chest help to loosen the mucus and clear it from the lungs. This reduces the risk of developing lung infections. Mucus-thinning drugs may also be administered.
- Careful monitoring is required to identify lung infections. Antibiotics are often prescribed to help manage and prevent infections.
- Supplementing the diet with enzymes normally produced by the pancreas to help break down food can help patients whose pancreas is blocked with mucus.
- In 2022, a new treatment called Kaftrio became available to more patients with cystic fibrosis on the NHS. Kaftrio is a ‘triple therapy’ with three active substances that work together to increase the number of CFTR proteins on the cell surface, while improving the activity of the defective CFTR protein. These actions combine to make lung mucus and digestive juices less thick, helping to relieve symptoms of the disease.
- Quality of life and life expectancy for people with cystic fibrosis have improved dramatically over the past few decades, thanks to increased understanding of the condition and new treatments becoming available.