
Post-sequencing: putting the sequence back together
Image credit: adapted from Mazza and Castellana, DOI: a6020309

How do you put a genome back together after sequencing?
This is part 2 in our series looking at what happens after DNA is sequenced. You might want to start with part 1, looking at the first step of quality control.
- After DNA sequencing is complete, the fragments of DNA that come out of the machine are all jumbled up.
- After a quality control check, the next step is to take the pieces of the genome and put them back together – like a jigsaw puzzle.
- This is our alignment and assembly step.
What’s the challenge?
- The technology of DNA sequencing is not 100% accurate, so it’s likely that there are errors in the DNA sequence that is produced.
- To account for these errors, each base in the genome is sequenced many times – a process called coverage. For example, 30-fold coverage means each base has been sequenced 30 times.
- 30- to 50-fold coverage is currently the standard used when sequencing human genomes to a high level of accuracy. The higher the coverage, the higher the confidence that the final sequence is correct.
- To put this into perspective, once a human genome has been fully sequenced, we have around 100 giga-bases (100,000,000,000 bases) of sequence data.
- Like the pieces of a jigsaw puzzle, these DNA reads are jumbled up. We need to piece them together and put them in the correct order to assemble the genome sequence.
What do we need to do?
- We need to put the pieces together in the correct order to construct the complete genome sequence and identify any areas of interest.
- This is done using processes called alignment and assembly.
- Alignment compares the new DNA sequence to existing DNA sequences, looking for any similarities or discrepancies between them and then arranging to show these features.
- Assembly takes a large number of DNA reads, looking for areas in which they overlap with each other and then gradually piecing together the ‘jigsaw’, in an attempt to reconstruct the original genome. This is primarily carried out for de novo sequences – when the genome of an organism is sequenced for the first time.
De novo sequencing
Producing a physical gene map for a de novo sequence
- De novo sequencing is when the genome of an organism is sequenced for the first time.
- In de novo assembly, there is no existing reference genome sequence for that species to use as a template for the assembly of its genome sequence.
- If you know that the new species is very similar to another species that does have a reference genome, it’s possible to assemble the sequence using a similar genome as a guide.
- To help assemble a de novo sequence a physical gene map can be developed before sequencing to highlight the ‘landmarks’ so the scientists know where sections of DNA are located in relation to each other.
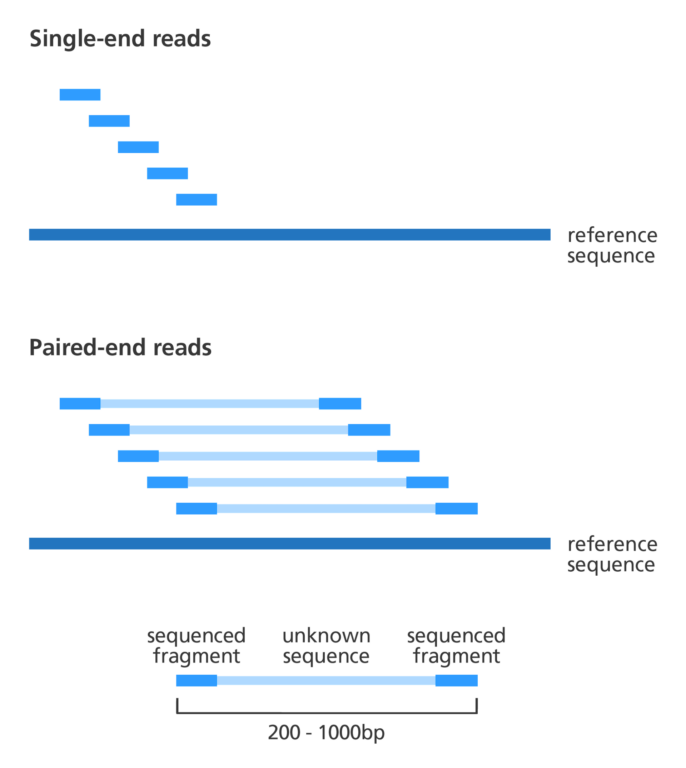
- Producing a gene map can be an expensive process, so some assembly programmes rely on data consisting of a mix of single and paired-end reads (see illustration below):
- Single reads are where one end or the whole of a fragment of DNA is sequenced. These sequences can then be joined together by finding overlapping regions in the sequence to create the full DNA sequence.
- Paired-end reads are where both ends of a fragment of DNA are sequenced. The distance between paired-end reads can be anywhere between 200 and several thousand bases. The key advantage of paired-end reads is that scientists know how far apart the two ends are. This makes it easier to assemble them into a continuous DNA sequence. Paired-end reads are particularly useful when assembling a de novo sequence as they provide long-range information that you wouldn’t otherwise have in the absence of a gene map.
De novo sequencing: assembly and alignment
- Assembly of a de novo sequence begins with many short sections or ‘reads’ of DNA. These reads are compared to each other and those sharing the same DNA sequence are grouped together.
- From here, they are assembled into progressively larger sections to form long contiguous (together in sequence) sequences called ‘contigs’.
- These contigs can then be grouped together with information taken from other technologies to provide clues for how to stitch the contigs together and roughly how far apart to place them, even if the sequence in between is still unknown. This is called “scaffolding”.
- The assembly can be further refined by ordering the individual scaffolds into chromosomes, often using a physical gene map as a guide.
- The resulting assembly is then fed on to the annotation, which identifies where the genes and other features in the sequence start and stop.
- As sequencing technologies have advanced, so too has the speed with which we can reassemble genomes. However, assembling a de novo genome where there is no reference to compare to remains a computer-intensive job.
Resequencing
- Resequencing is when the genome being sequenced is known to be from a species that has been sequenced before and so a reference genome is available.
- Resequencing is a term that can be used to describe two distinct processes: to improve the quality of the existing sequence, or to sequence the genome of another individual.
Resequencing to improve the quality of the available sequence
- The Human Genome Project, which was completed in 2003, provided a near-fully assembled sequence of the human genome.
- Since then, scientists have been working to produce a reference sequence of a higher quality and accuracy.
- As a result, the human reference genome has been vastly improved since 2003, with scientists correcting errors, rearranging the order of the individual contigs and filling any remaining gaps in the sequence.
Resequencing for comparison
- Another use of resequencing is when we sequence the genome of an individual from a species that we already have a reference genome for and know a bit about. We can then compare the new genome sequence with that of the reference and find out how they vary.
- For example, if there is a base-pair change in the new genome that isn’t present in the reference genome, it may give a clue as to the genetic origin of a particular trait or disease.
- We’ll go into more detail on this in part 4 of this series, looking at the comparison of genomes.