
Using phylogenetics to track disease outbreaks
By Juliana Cudini, (Previous) PhD Student at Wellcome Sanger Institute.

Phylogenetics is the study of the evolutionary relationships between organisms, based on their genomes - now used to track down disease outbreaks globally.
Key terms
Epidemic
A disease outbreak that's rapidly spreading in a limited region.
Pandemic
A disease outbreak that's actively spreading across the world.
What’s an outbreak?
An outbreak occurs when a disease (often caused by an infectious parasite, virus, or bacterium) spreads through a population. The term itself is somewhat of a ‘catch-all’ phrase that covers a range of scenarios. An outbreak can occur in very small populations, like a school class or a hospital ward, or spread to larger populations like entire cities or states.
Once an outbreak reaches a certain size, it is often given a new name – an epidemic. If an epidemic spreads to multiple countries or continents, it is upgraded to a pandemic. If an outbreak sticks around in a population for a long period of time, it becomes endemic. The suffix ‘-demic’ stems from the Greek word ‘demos’, meaning public, or population. The prefix ‘pan-’ means all, where the prefix ‘epi-’ means upon, or near.
Getting to the root of the matter
When you catch a cold, you often reconstruct the events of the previous few days to work out a likely culprit for the source of your infection, like the person who sneezed on you on the bus yesterday, or the slimy railing you held onto when climbing the stairs this morning. Understanding where we catch infections helps us avoid getting sick in the same way again. This is why, when a disease outbreak is first detected, scientists often scramble to track down where it might have come from, so we can stop it before it spreads any further.
In the same way you examined the events leading up to your first signs of an itchy throat when you catch a cold, scientists build an outbreak report, or an epidemiological history, of the cases detected so far and where they were found, to try and understand where they may have come from. This idea is the foundation for ‘Track and Trace’, used in the Covid-19 pandemic of the 2020s, which attempts to prevent future cases of disease from developing by tracking new cases and limiting their exposure with others.
Tracking and tracing using phylogenetic trees
While Track and Trace works by gathering information regarding who has contacted whom in small populations, this approach gets very difficult when an epidemic or pandemic spreads to millions of people. We can’t possibly reconstruct the contact history of hundreds of thousands of people with a disease. Instead, in these cases, we use genome sequencing to do the hard work for us.
Pathogens like viruses, bacteria, or parasites, change their genome sequence over time as they reproduce and pass from person to person. These changes, or mutations, serve as a trail of breadcrumbs that scientists can follow to track and trace a pathogen as it spreads through very large numbers of people. This is done by placing the genome sequence from each case tested onto a phylogenetic tree. Sequences that are similar will sit near to each other on the tree, implying they shared a common source of infection, even if very distantly.
Sequences that acquire mutations that make them dissimilar from those already observed can result in new branches being drawn on the tree. These are often termed variants, and they are carefully monitored for changes in disease severity or transmission. The more sequences we add to a phylogenetic tree, the better we understand where an outbreak originated and where it might go, which is why it is important to test as many cases as possible.
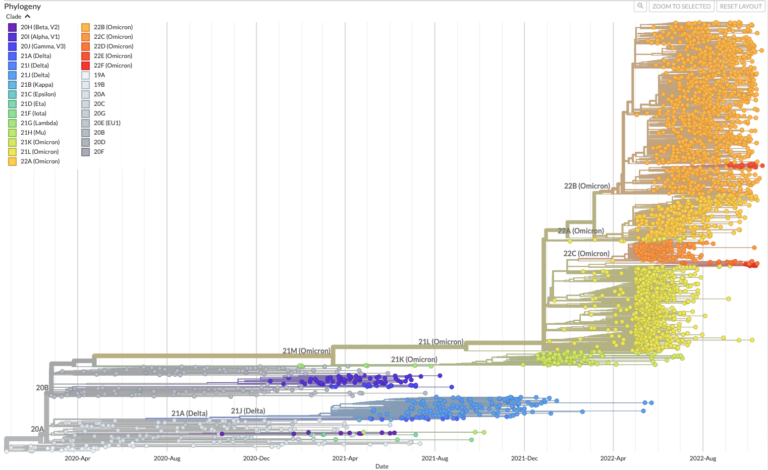
Using phylogenetic trees to predict the future
Using phylogenetic trees, we can map pathogens such as viruses, bacteria and parasites in the same way that we map related animal and plant species together, based on characteristics that they share. Pathogens with similar characteristics may be recognised as the same by the immune system, meaning that if you catch one of these viruses, your immune system is likely to be able to recognise and fight off any other member of that group you may come in contact with. This is called ‘cross-reactivity’.
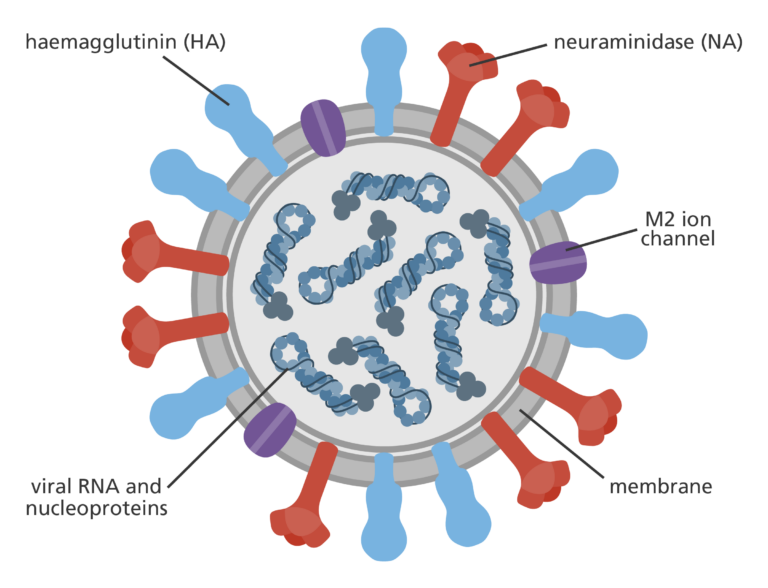
In the case of influenza A viruses – the virus behind the seasonal flu – tracking the size and location of these groupings around the world helps researchers predict which type is most likely to cause the largest number of cases during the next flu season. These types often centre around 2 proteins that sit on the surface of the virus; hemagglutinin (HA) and neuraminidase (NA). The virus uses these two proteins to get into cells and establish infection. The classification system for influenza A viruses depends on what combination of these two proteins a virus is carrying based on its sequence, often denoted as an ‘H type’ and ‘N type’. The swine flu pandemic in 2009 and the 1918 influenza pandemic (also known as Spanish Flu), for example, were both caused by an H1N1 virus, meaning they contained H type 1 and N type 1 proteins.
When flu vaccines are made, researchers observe which H-N combinations are currently circulating and predict which ones are the most likely to cause outbreaks during the next flu season. Since the viruses in these types share common characteristics, the vaccines developed cross-react to protect against all other influenza A viruses in that group. When predictions based on the influenza phylogeny are correct, the flu jab can be highly effective in stopping outbreaks before they even get a chance to take off.
Article written by Juliana Cudini, PhD Student at Wellcome Sanger Institute